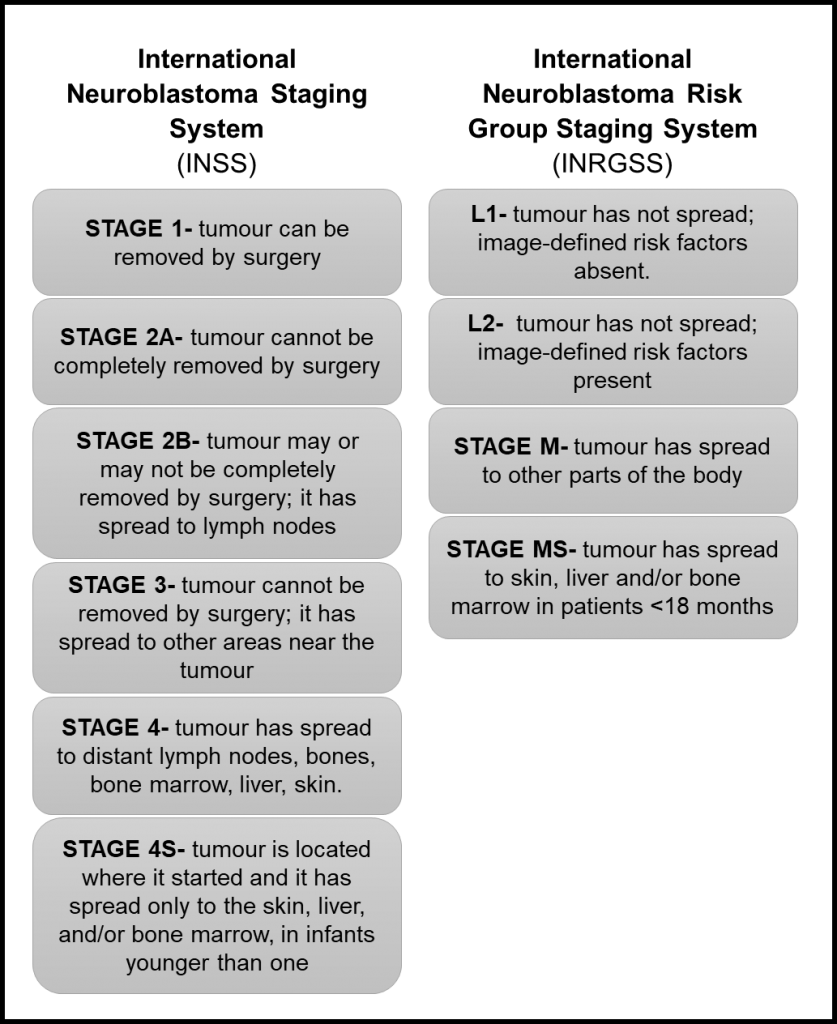
The determination of the tumour stage is an important step after a neuroblastoma diagnosis. The stage of neuroblastoma is determined depending on tumour location and if it has spread to other parts of the body. This will guide risk group assignment and treatment choice.
The first staging system for neuroblastoma, the International Neuroblastoma Staging System (INSS), was developed in 1986 and is based on the pathological evaluation of the tumour after a removal surgery. In 2005, The International Neuroblastoma Risk Group Staging System (INRGSS) started to be used. This system is based on tumour images before any surgery. Therefore, it is based on image-defined risk factors to determine the tumour stage (see table below). It also uses clinical, pathologic, and genetic markers to determine the risk groups, which can be low-risk, intermediate-risk, or high-risk.
Reference: Neuroblastoma – Childhood: Stages and Groups, Cancer.net.
Recently, the Children’s Oncology Group (COG), a clinical trial group dedicated to paediatric cancer research revised the classification system they use to determine tumour stage for enrolment in clinical trials1. Previously, they have been defining the tumour stage based on the INNS system. Now they proposed a revised classification that takes into account the INRGSS and chromosomal alterations.
Key clinical and biological factors used in the neuroblastoma risk classification include age at diagnosis, disease stage, tumour tissue appearance under a microscope (histology), the status of the gene MYCN that affects tumour growth, the amount of DNA in a tumour cell (called tumour cell ploidy), and alterations in the DNA.
They analyse the outcome of almost 5,000 patients to define risk groups based on the INRGSS, using alterations in the DNA of tumour cells as a biomarker and considering current therapy modalities. In general, they found that the correlation of stages between systems is not exact. However, the differences in survival were minimal when comparing staging systems, which corroborates the use of the revised version.
In general, the new version classifies L1 and L2 tumours as low risk, except for L1 tumours with alteration in the gene MYCN and that cannot be removed by surgery, which is high-risk. For L2 tumours, MYCN status and age can be used to evaluate prognosis. Stage M tumours can be classified as high risk or intermediate-risk depend on age, MYCN status and DNA alterations. In conclusion, low-risk groups have excellent outcomes with any or limited therapy, the intermediate-risk group have very good outcomes and high-risk groups have inferior outcomes despite therapy.
This new version of the COG classifier will provide a uniformization of patient risk classification for clinical trials, ultimately enabling the comparison between different trials.
Written by Luiza Erthal
Reference:
1. Irwin, M. S. et al. Revised Neuroblastoma Risk Classification System: A Report From the Children’s Oncology Group. J. Clin. Oncol. JCO.21.00278 (2021)
This post is dedicated to parents of children with neuroblastoma. Some parents asked about DFMO – a re-purposing drug. In this post, I tried to collect and summarize information available from academic sources.
Q1: What is DFMO?
Difluoromethylornithine (DFMO, Eflornithine) is an anti-protozoan drug. It was originally developed and FDA approved for the treatment of Trypanosoma brucei gambiense encephalitis (“African sleeping sickness”). DFMO permanently binds to ornithine decarboxylase (ODC), an important enzyme in polyamine metabolism, and prevents the natural substrate ornithine from entering the active site.
By inhibiting ODC, DFMO reduces cellular polyamines and inhibits cell growth and proliferation of actively dividing cells, thus making DFMO an attractive candidate for cancer therapy. In neuroblastoma, a positive regulation of all aspects of polyamine metabolism by MYCN was reported (revived by Bassiri 2015, Gamble 2012). So, it is believed that MYCN amplified neuroblastomas would most benefit of the drug.
Q2: How intense is basic science behind DFMO in neuroblastoma?
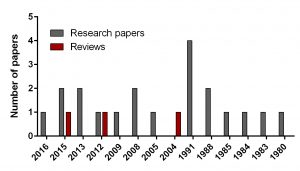
Figure 1. Breakdown of papers covering DFMO in neuroblastoma
To find out the intensity of basic science on DFMO in neuroblastoma search for ‘difluoromethylornithine/DFMO/Eflornithine’ and ‘neuroblastoma’ was run in PubMed, a web-based resource with 26 million citations for biomedical literature from MEDLINE, life science journals, and online books. The search returned 23 papers including 3 reviews and 20 primary research reports published from 1980 to present.
In comparison, I did another search for a novel drug Unituxin (dinutuximab) approved by FDA in 2015. It is monoclonal antibody against the glycolipid disialoganglioside GD2, a biomarker specific for neuroblastoma. Search for ‘anti-GD2 antibody’ and ‘neuroblastoma’ returned 181 papers including 25 reviews and 156 primary articles for the same period.
Q3: Is DFMO in cancer clinical trials?
“ClinicalTrials.gov is a Web-based resource that provides patients, their family members, health care professionals, researchers, and the public with easy access to information on publicly and privately supported clinical studies on a wide range of diseases and conditions. The Web site is maintained by the National Library of Medicine (NLM) at the National Institutes of Health (NIH).
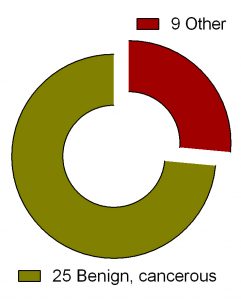
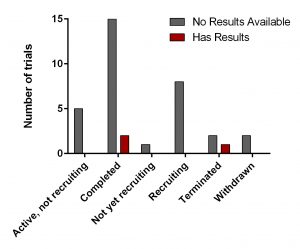
Figure 2. Clinical trials of DFMO in various health conditions.
Search for ‘difluoromethylornithine/DFMO/Eflornithine’ in ClinicalTrials.gov returned 36 registered trials across different health conditions.Two of these were withdrawn, the breakdown for the rest 34 is as follows: Adenomatous Polyp (1), Anaplastic Astrocytoma/Recurrent Anaplastic Astrocytoma (1), Bladder Cancer (1), Cervical Cancer/Precancerous Condition (1), Colorectal Cancer (3), Esophageal Cancer (1), Familial Adenomatous Polyposis (1), Gastric Cancer/Gastric Intestinal Metaplasia (1), Hirsutism (2), Human African Trypanosomiasis (5), Neuroblastoma (7), Non-melanomatous Skin Cancer/Precancerous/Nonmalignant Condition (4), Post-solid Organ Transplant/Skin Neoplasms (1), Precancerous Condition (1), Prostate Cancer (2), Pseudofolliculitis Barbae (1), Type 1 Diabetes (1) (Fig. 2). To see full details of 34 trials please click at this Table.
Figure 3. DFMO clinical trials from ClinicalTrials.gov
All of them have various statuses (Fig. 3) as well as study design. Importantly, 30 out of 34 studies are focused on safety and efficacy of this drug. Vast majority of studies of DFMO in adult cancers/benign conditions are randomized (16/18 or 89%). Randomization in assignment of patients in studied groups (control and new drug/combination) helps minimize researcher’s bias when comparing effect of the new treatment vs current/no treatment. All trials of DFMO in neuroblastoma are not randomized. Instead, studies use a single group assignment.
Three trails have been either completed/terminated and published results are available at ClinicalTrials.gov (NCT01059071, NCT00033371. NCT00118365).
Q4: What about clinical trials of DFMO in neuroblastoma?
The trial NCT01059071 was a Phase 1 clinical trial. A phase I clinical trial tries to find out whether a new treatment/drug is safe, what its side effects are, the best dose of the new treatment, if the treatment shrinks the cancer.
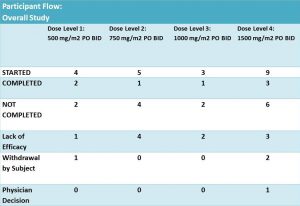
Twenty one patients were enrolled and eligible for treatment with DFMO and DFMO + etoposide. These patients were assigned into 4 groups of different DFMO doses (Fig. 4). The treatment was in cycles of 21 days. Cycle 1 – DFMO only followed by cycle 2 – combined treatment of DFMO+etoposide (14 days) and DFMO only (the last 7 days).
Figure 4. Table was adopted from results section of the clinical trial NCT01059071
According to results of the trial: 14 patients did not complete the treatment due to different reasons. It was not clear what stage/cycle they left the trial.
As mentioned earlier this study used a single group assignment and a design called ‘3+3’. This design is straightforward and safe. Briefly, it means that for a dose (X) of the drug, 6 patients are selected. Of these, 3 receive the dose X and are monitored for a period of time. If no adverse effects are registered in these 3, then another new 3 patients start the same treatment. The effect of the drug is evaluated on the patent’s health condition before-, during – the treatment and after its completion. This approach is often used in vaccine tests and dose escalation methods in Phase I cancer clinical trials. This type of study can answer mainly two questions: 1) whether the tested drug is safe to use and 2) what doses are safe? The main drawbacks of this design are
Many patients treated at doses below therapeutic effect
Slow dose increase
Uncertainty about the recommended phase II dose (RP2D)
Only the result from the current dose is used for determining the dose of next cohort of patients. Information on other doses is ignored
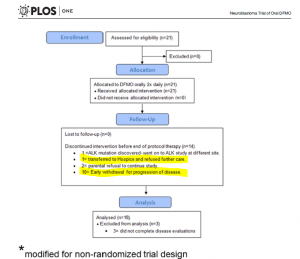
Figure 5 is adopted from Sholler GL et al, PLoS One. 2015 May 27;10(5):e0127246.
Q6: What are main findings of the clinical trial NCT01059071?
The overflow of the study is presented in Fig 5 providing additional information on those who did not complete the trial. Out of 14 participants, disease has progressed in 11 patients – it is 52% of the enrolled participants. Authors highlighted that this phase I study was not designed to evaluateanti-tumour efficacy of DFMO. But tumour response and clinical response were monitored during the study.
According to the paper, 21 patients received at least one dose of DFMO only (Cycle 1, 21 days). During this cycle, 3 patients were withdrawn. All of them were assessed for safety of DFMO.
Eighteen of them completed cycle 1 and continued treatment with DFMO+etoposide for another 4 cycles followed with DFMO only therapy for a number of cycles. Their clinical response data were examined for efficacy of DFMO alone.
Three out of 21 participating patients in this clinical trial remain alive and disease free between 2–4.5 years after starting DFMO.
Authors concluded that
DFMO doses of 500-1500mg/m2/day are safe and well tolerated in children with relapsed NB
Research and review papers covering DFMO in neuroblastoma:
Evageliou NF, Haber M, Vu A, Laetsch TW, Murray J, Gamble LD, Cheng NC, Liu K, Reese M, Corrigan KA, Ziegler DS, Webber H, Hayes CS, Pawel B, Marshall GM, Zhao H, Gilmour SK, Norris MD, Hogarty MD. Polyamine Antagonist Therapies Inhibit Neuroblastoma Initiation and Progression. Clin Cancer Res. 2016 Sep 1;22(17):4391-404. doi: 10.1158/1078-0432.CCR-15-2539.
Bassiri H, Benavides A, Haber M, Gilmour SK, Norris MD, Hogarty MD. Translational development of difluoromethylornithine (DFMO) for the treatment of neuroblastoma. Transl Pediatr. 2015 Jul;4(3):226-38. doi: 10.3978/j.issn.2224-4336.2015.04.06. Review.
Saulnier Sholler GL, Gerner EW, Bergendahl G, MacArthur RB, VanderWerff A, Ashikaga T, Bond JP, Ferguson W, Roberts W, Wada RK, Eslin D, Kraveka JM, Kaplan J, Mitchell D, Parikh NS, Neville K, Sender L, Higgins T, Kawakita M, Hiramatsu K, Moriya SS, Bachmann AS. A Phase I Trial of DFMO Targeting Polyamine Addiction in Patients with Relapsed/Refractory Neuroblastoma. PLoS One. 2015 May 27;10(5):e0127246. doi: 10.1371/journal.pone.0127246.
Lozier AM, Rich ME, Grawe AP, Peck AS, Zhao P, Chang AT, Bond JP, Sholler GS Targeting ornithine decarboxylase reverses the LIN28/Let-7 axis and inhibits glycolytic metabolism in neuroblastoma. Oncotarget. 2015 Jan 1;6(1):196-206.
Samal K, Zhao P, Kendzicky A, Yco LP, McClung H, Gerner E, Burns M, Bachmann AS, Sholler G. AMXT-1501, a novel polyamine transport inhibitor, synergizes with DFMO in inhibiting neuroblastoma cell proliferation by targeting both ornithine decarboxylase and polyamine transport. Int J Cancer. 2013 Sep 15;133(6):1323-33. doi: 10.1002/ijc.28139.
Koomoa DL, Geerts D, Lange I, Koster J, Pegg AE, Feith DJ, Bachmann AS. DFMO/eflornithine inhibits migration and invasion downstream of MYCN and involves p27Kip1 activity in neuroblastoma. Int J Oncol. 2013 Apr;42(4):1219-28. doi: 10.3892/ijo.2013.1835.
Gamble LD, Hogarty MD, Liu X, Ziegler DS, Marshall G, Norris MD, Haber M. Polyamine pathway inhibition as a novel therapeutic approach to treating neuroblastoma. Front Oncol. 2012 Nov 16;2:162. doi: 10.3389/fonc.2012.00162. Review
Passariello CL, Gottardi D, Cetrullo S, Zini M, Campana G, Tantini B, Pignatti C, Flamigni F, Guarnieri C, Caldarera CM, Stefanelli C. Evidence that AMP-activated protein kinase can negatively modulate ornithine decarboxylase activity in cardiac myoblasts. Biochim Biophys Acta. 2012 Apr;1823(4):800-7. doi: 10.1016/j.bbamcr.2011.12.013.
Rounbehler RJ, Li W, Hall MA, Yang C, Fallahi M, Cleveland JL. Targeting ornithine decarboxylase impairs development of MYCN-amplified neuroblastoma. Cancer Res. 2009 Jan 15;69(2):547-53. doi: 10.1158/0008-5472.CAN-08-2968.
Koomoa DL, Yco LP, Borsics T, Wallick CJ, Bachmann AS. Ornithine decarboxylase inhibition by alpha-difluoromethylornithine activates opposing signaling pathways via phosphorylation of both Akt/protein kinase B and p27Kip1 in neuroblastoma. Cancer Res. 2008 Dec 1;68(23):9825-31. doi: 10.1158/0008-5472.CAN-08-1865.
Hogarty MD, Norris MD, Davis K, Liu X, Evageliou NF, Hayes CS, Pawel B, Guo R, Zhao H, Sekyere E, Keating J, Thomas W, Cheng NC, Murray J, Smith J, Sutton R, Venn N, London WB, Buxton A, Gilmour SK, Marshall GM, Haber M. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008 Dec 1;68(23):9735-45. doi: 10.1158/0008-5472.CAN-07-6866.
Wallick CJ, Gamper I, Thorne M, Feith DJ, Takasaki KY, Wilson SM, Seki JA, Pegg AE, Byus CV, Bachmann AS. Key role for p27Kip1, retinoblastoma protein Rb, and MYCN in polyamine inhibitor-induced G1 cell cycle arrest in MYCN-amplified human neuroblastoma cells. Oncogene. 2005 Aug 25;24(36):5606-18.
Bachmann AS. The role of polyamines in human cancer: prospects for drug combination therapies. Hawaii Med J. 2004 Dec;63(12):371-4. Review
Chen ZP, Chen KY. Differentiation of a mouse neuroblastoma variant cell line whose ornithine decarboxylase gene has been amplified. Biochim Biophys Acta. 1991 Dec 3;1133(1):1-8.
Piacentini M, Fesus L, Farrace MG, Ghibelli L, Piredda L, Melino G. The expression of “tissue” transglutaminase in two human cancer cell lines is related with the programmed cell death (apoptosis). Eur J Cell Biol. 1991 Apr;54(2):246-54.
Melino G, Piacentini M, Patel K, Annicchiarico-Petruzzelli M, Piredda L, Kemshead JT. Retinoic acid and alpha-difluoromethylornithine induce different expression of neural-specific cell adhesion molecules in differentiating neuroblastoma cells. Prog Clin Biol Res. 1991;366:283-91.
Stephanou A, Knight RA, De Laurenzi V, Melino G, Lightman SL.Expression of pre-opiomelanocortin (POMC) mRNA in undifferentiated and in vitro differentiated human neuroblastoma cell lines. Prog Clin Biol Res. 1991;366:173-80.
Melino G, Farrace MG, Ceru’ MP, Piacentini M. Correlation between transglutaminase activity and polyamine levels in human neuroblastoma cells. Effect of retinoic acid and alpha-difluoromethylornithine. Exp Cell Res. 1988 Dec;179(2):429-45.
Chen KY, Dou QP. NAD+ stimulated the spermidine-dependent hypusine formation on the 18 kDa protein in cytosolic lysates derived from NB-15 mouse neuroblastoma cells. FEBS Lett. 1988 Mar 14;229(2):325-8.
Karvonen E, Andersson LC, Pösö H. A human neuroblastoma cell line with a stable ornithine decarboxylase in vivo and in vitro. Biochem Biophys Res Commun. 1985 Jan 16;126(1):96-102.
Pösö H, Karvonen E, Suomalainen H, Andersson LC. A human neuroblastoma cell line with an altered ornithine decarboxylase. J Biol Chem. 1984 Oct 25;259(20):12307-10.
Chen KY, Nau D, Liu AY. Effects of inhibitors of ornithine decarboxylase on the differentiation of mouse neuroblastoma cells. Cancer Res. 1983 Jun;43(6):2812-8.
Chapman SK. Antitumor effects of vitamin A and inhibitors of ornithine decarboxylase in cultured neuroblastoma and glioma cells. Life Sci. 1980 Apr 21;26(16):1359-66. No abstract available.
Ok. Now, when the stress of the presentation is over, I am happy to share new technologies used during the SIOP2016. As I mentioned yesterday, my work was selected for e-poster presentation. It looked this way:
This is e-poster station, where anyone can look up all posters displayed during the meeting.
It is definitely a step forward. Anyone can look up any poster, listen to a commentary recorded by the author, zoom in and out and send a request/comment to the author. It looks cool and trendy. Though, you can feel invisible as no physical copy displayed in a designated area. No crowds of poster presenters and judges. No waiting faces desperate to share their study…
The actual Poster Discussion session was a traditional presentation when my poster was up on the big screen, I had 8 minutes to convince the audience navigating through figures. This session was late and no many attendees survived to come and challenge your statements. Nevertheless, it was enjoyable experience. 🙂
Currently, the only popular trend in science is to publish only those results that look as a breakthrough or display statistically significant data. Obsession with positive outcome leads to discontinuation and non-publishing the all other data which don’t meet the requirements. As a result researchers and public did not know mistakes, efforts and drilling details of non-positive studies, so they have no opportunity to review this data, refine the research hypothesis and technical performance. This leads to waste of time and funding money.
The very recent study by Natalie Pica, MD, PhD, and Florence Bourgeois MD, MPH from the Department of Pediatrics at Harvard Medical School and Boston Children’s Hospital, both in Massachusetts has again confirmed the problem. The researchers carried out a retrospective, cross-sectional study of childhood randomized controlled trials and published their findings in Pediatrics(DOI: 10.1542/peds.2016-0223). They collected information from all trials that were registered ClinicalTrials.gov from 2008 to 2010, then searched scientific publications based on the trials. They also verified final status of the selected trials (completed or discontinued) by the end of 2012. If researchers found no publication, they contacted investigators and sponsors associated with trials to clarify the issue.
The main findings were:
19% of 559 trials were discontinued early representing approximately 8369 children. The most common reason for discontinuation – difficulty with patient accrual (37%).
30 % of the 455 completed trials were not published, representing 69 165 children participants.
Only 42 unpublished trials posted results on ClinicalTrials.gov.
Trials were less likely to be dropped if they were funded by industry.
Trials funded by industry were more than twice as likely to result in nonpublication and a longer mean time to publication when compared with trials sponsored by academia.
Researchers concluded that “withdrawal and nonpublication were common, resulting in thousands of children exposed to interventions that did not lead to informative or published findings. Trial funding source was an important determinant of these outcomes, with both academic and industry sponsors contributing to inefficiencies.” (Pica N & Bourgeois F, 2016, e 20160223)
Pica N & Bourgeois F, Discontinuation and Nonpublication of Randomized Clinical Trials Conducted in Children. PEDIATRICS V 138(3) 2016:e 20160223 Access to the study can be found here: